2020年10月15日,上海交通大学生物医学工程学院李力课题组和武汉大学生命科学学院吴旻课题组在国际著名期刊Hepatology杂志上发表了题为Deficiency of histone methyltransferase SETD2 in liver leads to abnormal lipid metabolism and hepatocarcinoma的研究工作,证实了SETD2缺失促进肝癌(HCC)发生,并阐明了SETD2在肝癌中调节胆固醇稳态和c-Jun/AP1信号转导的作用机制。
肝癌是世界常见恶性肿瘤,在亚洲尤其是我国发病率极高。肝脏在体内胆固醇代谢平衡中发挥极重要作用。肝癌的发生发展是一个非常复杂的生物学过程,受到机体的系统性调控。肝癌使肝脏代谢功能受到极大的损伤,导致包括胆固醇代谢在内的许多代谢过程出现病理性变化。然而,胆固醇代谢变化的调控机理在肝癌发生发展中的作用还不清楚。
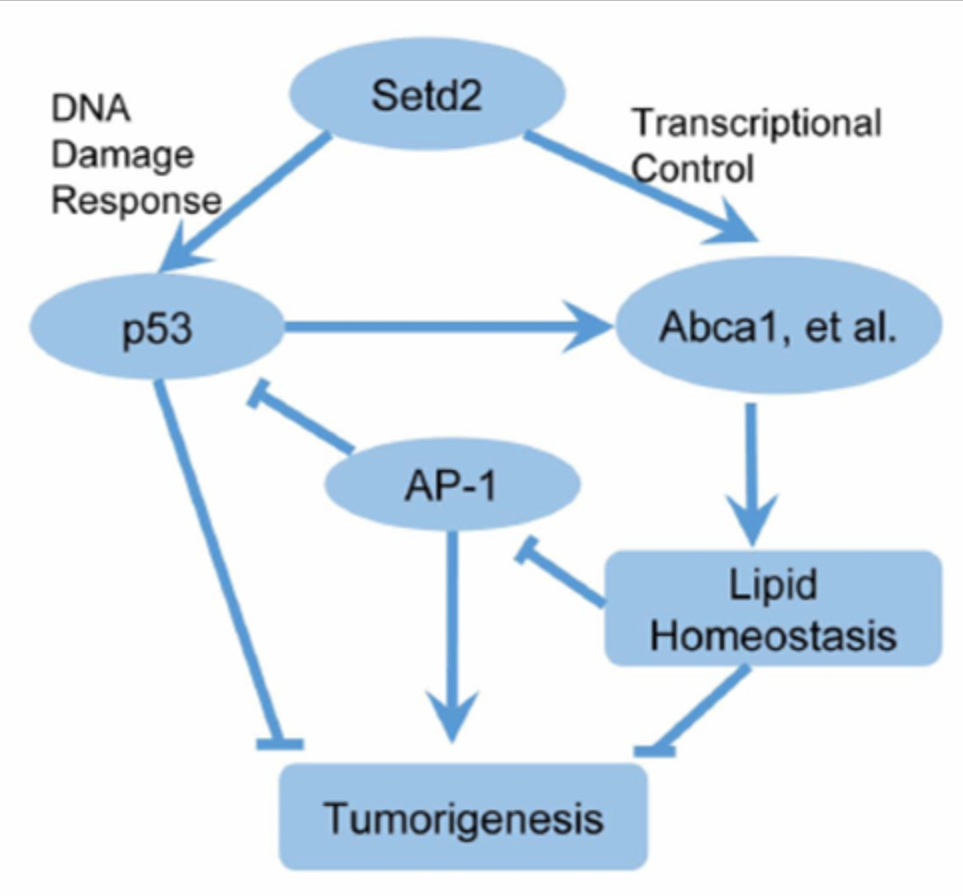
组蛋白甲基转移酶SETD2(SET domain-containing protein 2)催化的H3K36me3是从酵母到哺乳动物最保守的表观遗传标记之一。SETD2在多种癌症中经常发生突变。李力副研究员在2014年建立了Setd2条件性基因敲除小鼠模型并在生殖、发育和癌症等领域展开广泛的功能研究(Zuo et al, J Bio Chem, 2018;Wang et al, PLoS Biol, 2018;Xu et al, Nat Genet, 2019;Ji et al,Nat Commun,2019;Niu et al,Gut,2020)。合作组由此展开进一步研究,利用肝脏特异性Setd2缺失模型,发现Setd2缺乏足以引发自发性肝癌。同时,Setd2缺失显著增加了DEN诱导的HCC模型的肿瘤数量和肿瘤大小。

进一步研究表明,Setd2不仅通过调节DNA损伤反应抑制HCC,而且通过调节肝脏脂质代谢来抑制HCC。Setd2缺乏可下调H3K36me3胆固醇外流基因的富集和表达,引起脂质积聚。高脂饮食促进Setd2基因缺陷小鼠脂质积累,促进肝癌的发生。ChIP-Seq分析显示,Setd2缺失可诱导肝脏c-Jun/AP-1激活,这是由脂质累积引起的。c-Jun作为HCC的癌基因,通过抑制Setd2缺陷细胞中的p53发挥作用。本研究首次揭示了Setd2在肝癌中的作用,揭示了其调节胆固醇稳态和c-Jun/AP1信号转导的机制,深入了对其病理机制的认识,为肝癌靶向治疗提供了新的理论依据。
武汉大学李雪晶为该论文的第一作者;吴旻教授和李力副研究员为该论文的共同通讯作者。该研究课题得到了国家科技部,国家自然科学基金委和上海市科委的资助。
原文链接: https://doi.org/10.1002/hep.31594









